


Bit raw-data heavy, but for those of us with damaged knees, some hope...
Int J Med Sci 2009; 6(6):312-321. doi:10.7150/ijms.6.312
Research Paper
Safety and efficacy of undenatured type II collagen in the treatment of osteoarthritis of the knee: a clinical trial
David C. Crowley[SUP]1[/SUP], Francis C. Lau[SUP]2[/SUP], Prachi Sharma[SUP]1[/SUP], Malkanthi Evans[SUP]1[/SUP], Najla Guthrie[SUP]1[/SUP], Manashi Bagchi[SUP]2[/SUP], Debasis Bagchi[SUP]2,3[/SUP], Dipak K. Dey[SUP]4[/SUP], Siba P. Raychaudhuri[SUP] 5,6, [/SUP]
[/SUP]
1. KGK Synergize Incorporated, London, ON, Canada
2. Department of Research and Development, InterHealth Research Center, Benicia, CA, USA
3. Department of Pharmacology and Pharmaceutical Sciences, University of Houston College of Pharmacy, Houston, TX, USA
4. Department of Statistics, University of Connecticut, Storrs, CT, USA
5. Department of Medicine, Division of Rheumatology, Allergy and Immunology, School of Medicine, University of California Davis, Davis, CA, USA
6. VA Medical Center Sacramento, Hospital Way, Mather, CA, USA
How to cite this article:
Crowley DC, Lau FC, Sharma P, Evans M, Guthrie N, Bagchi M, Bagchi D, Dey DK, Raychaudhuri SP. Safety and efficacy of undenatured type II collagen in the treatment of osteoarthritis of the knee: a clinical trial. Int J Med Sci 2009; 6(6):312-321. doi:10.7150/ijms.6.312. Available from http://www.medsci.org/v06p0312.htm
Abstract
Previous studies have shown that undenatured type II collagen (UC-II) is effective in the treatment of rheumatoid arthritis, and preliminary human and animal trials have shown it to be effective in treating osteoarthritis (OA). The present clinical trial evaluated the safety and efficacy of UC-II as compared to a combination of glucosamine and chondroitin (G+C) in the treatment of OA of the knee. The results indicate that UC-II treatment was more efficacious resulting in a significant reduction in all assessments from the baseline at 90 days; whereas, this effect was not observed in G+C treatment group. Specifically, although both treatments reduced the Western Ontario McMaster Osteoarthritis Index (WOMAC) score, treatment with UC-II reduced the WOMAC score by 33% as compared to 14% in G+C treated group after 90 days. Similar results were obtained for visual analog scale (VAS) scores. Although both the treatments reduced the VAS score, UC-II treatment decreased VAS score by 40% after 90 days as compared to 15.4% in G+C treated group. The Lequesne's functional index was used to determine the effect of different treatments on pain during daily activities. Treatment with UC-II reduced Lequesne's functional index score by 20% as compared to 6% in G+C treated group at the end of 90-day treatment. Thus, UC-II treated subjects showed significant enhancement in daily activities suggesting an improvement in their quality of life.
Keywords: undenatured type II collagen, osteoarthritis, glucosamine, chondroitin, WOMAC, visual analog scale, Lequesne's Functional Index
INTRODUCTION
Arthritis afflicts approximately 43 million Americans or approximately 16.6% of the US population. The two most common types of arthritis are osteoarthritis (OA) and rheumatoid arthritis (RA). OA of the knee and hip is a growing health concern and is the most common forms of arthritis (1-3). Pain and disease can range from very mild to very severe (3). Patients with OA have pain that typically worsens with weight bearing, including walking and standing, and improves with rest (4). Other symptoms include morning stiffness and gelling of the involved joint after periods of inactivity. Currently, OA affects nearly 21 million people in the United States, accounting for 25% of visits to primary care physicians, and half of all Non-Steroidal Anti-Inflammatory Drugs (NSAID) prescriptions. The diverse clinical patterns of OA are observed in approximately 10% of people older than 60 years thus compromising the quality of life of millions of Americans. In addition, OA costs the North American economy approximately $60 billion per year.
Current treatment of OA includes exercise, heat/cold therapy, joint protection, weight loss, physiotherapy/occupational therapy and medications (3-5). The most common medications include acetaminophen and NSAIDs. Although these drugs are effective for reducing pain associated with OA, they do not reverse the disease. In addition, there are considerable side effects associated with the use of these drugs. As a result, OA sufferers have turned to natural nutraceuticals to ease their pain and discomfort. These products are commonly used because they are well tolerated and considered safe. Nutraceuticals are defined as functional foods, natural products, or parts of food that provide medicinal, therapeutic, or health benefits, including the prevention or treatment of disease. Currently, glucosamine and chondroitin are the two most commonly used nutraceuticals in humans as well as in animals to alleviate pain associated with arthritis (6). However, recent randomized controlled trials and meta-analysis of these supplements have shown only small-to-moderate symptomatic efficacy in human OA (7). An emerging novel nutraceutical ingredient known as UC-II has received considerable attention in the treatment of OA. UC-II is a novel undenatured type II collagen derived from chicken sternum cartilage. Previous studies have shown that undenatured type II collagen is effective in the treatment of RA (8-11), and preliminary human (12) and animal (13) trials have shown it to be effective in treating OA. Obese-arthritic dogs given 4 mg or 40 mg daily dose of UC-II for 90 days showed significant declines in overall pain, pain during limb manipulation and lameness after physical exertion (14). Greater improvement was observed with the 40 mg dose. No adverse effects or significant changes in serum chemistry were noted. Following UC-II withdrawal for a period of 30 days, all dogs experienced a relapse of overall pain, exercise-associated lameness and pain upon limb manipulation. Studies have also shown that small doses of orally administered undenatured type II chicken collagen inhibit killer T-cell attack (15). The present clinical trial evaluated the safety and efficacy of UC-II in the treatment of the knee in OA patients.
Materials and Methods
Study Design
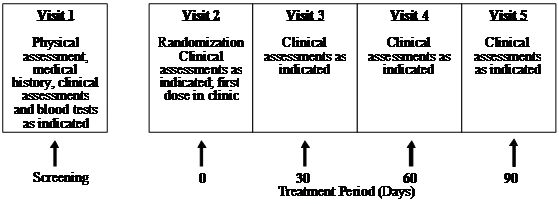
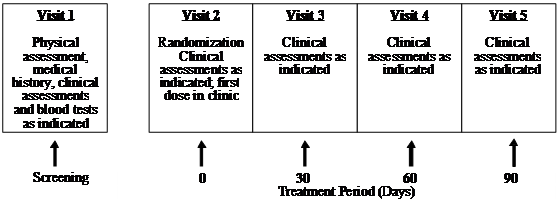
This clinical trial (Human Clinical Trial Approval #06UOHI) was managed by KGK Synergize Inc. (London, ON, Canada). The study was conducted at two sites: 1) KGK Synergize Inc., and 2) Corunna Medical Research (Corunna, ON, Canada). Figure 1 illustrates the study design while Table 1 lists the procedures and observations at each time point. Briefly, at screening (Visit 1) the consent form was discussed, signed and a complete physical examination was performed. Activity level, diet history, medication/supplement use and medical history were recorded. The VAS score, the WOMAC Index and Lequesne scores were obtained. Urine was collected for a pregnancy test for women of childbearing potential. A blood sample was taken for determination of uric acid, CBC count and differentiation, albumin, total protein, sodium, potassium, chloride, BUN, creatinine, ALT, AST, bilirubin, erythrocyte sedimentation rate (ESR) and rheumatoid factor. Upon review of blood test results, eligible subjects were instructed to get an X-ray of the affected knees to confirm diagnosis. A total of 52 subjects were recruited using the inclusion and exclusion criteria outlined in Table 2. At the first treatment visit (Visit 2), selected subjects were randomly assigned to receive UC-II (n = 26) or glucosamine HCl plus chondroitin sulfate (n = 26, G+C). On each test day (day 0, 30, 60, 90), subjects were required to come to the clinic for clinical assessment. The clinical assessments included WOMAC, Lequesne's functional index and 100-mm VAS pain scores. A subject treatment diary was completed by each patient throughout the study period to determine side effects, medication use, and product compliance.
Figure 1 UC-II clinical study design. The study was a two-site, randomized, double-blind study conducted in London, Ontario and Corunna, Ontario, Canada.
 (Click on the image to enlarge.)
(Click on the image to enlarge.)
Table 1 Schedule of observations and procedures
[TABLE="class: ivytablelight"]
<tbody>[TR]
[TH]Procedure[/TH]
[TH]Visit 1 Screening[/TH]
[TH]Visit 2 Day 0[/TH]
[TH]Visit 3 Day 30[/TH]
[TH]Visit 4 Day 60[/TH]
[TH]Visit 5 Day 90[/TH]
[/TR]
[TR]
[TD]Informed consent[/TD]
[TD]X[/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[/TR]
[TR]
[TD]Review inclusion/exclusion[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[/TR]
[TR]
[TD]Medical history including activity level and diet history[/TD]
[TD]X[/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[/TR]
[TR]
[TD]Physical examination[/TD]
[TD]X[/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[/TR]
[TR]
[TD]Biometric measurements: Weight, height*, heart rate and blood pressure.[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[/TR]
[TR]
[TD]Urine pregnancy test[/TD]
[TD]X[/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[/TR]
[TR]
[TD]Concomitant medications[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[/TR]
[TR]
[TD]Blood samples: Uric acid, CBC count and differentiation, albumin, total protein, sodium, potassium, chloride, BUN, creatinine, ALT, AST, bilirubin, ESR, rheumatoid factor[/TD]
[TD]X[/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[TD]X[/TD]
[/TR]
[TR]
[TD]WOMAC, VAS and Lequesne scores[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[/TR]
[TR]
[TD]X-ray[/TD]
[TD]X[/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[/TR]
[TR]
[TD]Randomization[/TD]
[TD][/TD]
[TD]X[/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[/TR]
[TR]
[TD]Blood sample: ALT, AST, bilirubin, albumin.[/TD]
[TD][/TD]
[TD][/TD]
[TD]X[SUP]†[/SUP][/TD]
[TD]X[SUP]†[/SUP][/TD]
[TD][/TD]
[/TR]
[TR]
[TD]Knee flexion, Time to walk 50m, Swelling in the knee joint, Time for climbing 10 steps[/TD]
[TD][/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[/TR]
[TR]
[TD]Physician's Global Assessment[/TD]
[TD][/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[/TR]
[TR]
[TD]Subject's Global Assessment[/TD]
[TD][/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[/TR]
[TR]
[TD]Investigational Product dispensed[/TD]
[TD][/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD][/TD]
[/TR]
[TR]
[TD]Subject Treatment Diary dispensed[/TD]
[TD][/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD][/TD]
[/TR]
[TR]
[TD]Investigational Product returned Compliance calculated[/TD]
[TD][/TD]
[TD][/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[/TR]
[TR]
[TD]Subject Treatment Diary returned[/TD]
[TD][/TD]
[TD][/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[/TR]
[TR]
[TD]Adverse Events[/TD]
[TD][/TD]
[TD][/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[/TR]
</tbody>[/TABLE]
* height was only measured at visit 1
[SUP]† [/SUP]If acetaminophen use was greater than 2 g/day for more than 7 days
Table 2 Inclusion and exclusion criteria
[TABLE="class: ivytablelight"]
<tbody>[TR]
[TH="align: center"]Inclusion Criteria[/TH]
[/TR]
[TR]
[TD]Males and females 40-75 years old[/TD]
[/TR]
[TR]
[TD]Females of childbearing potential must agree to use a medically approved form of birth control and have a negative urine pregnancy test result[/TD]
[/TR]
[TR]
[TD]Unilateral or bilateral OA of the knee for greater than 3 months (American College of Rheumatology criteria) confirmed by radiologist's report, i.e. X-rays showing osteophytes, joint space narrowing or subchondral bone sclerosis (eburnation)[/TD]
[/TR]
[TR]
[TD]Erythrocyte sedimentation rate (ESR) < 40 mm/hr[/TD]
[/TR]
[TR]
[TD]Moderate OA as indicated by Lequesne's functional index score of 4.5-7.5 after 7 day withdrawal of usual medications[/TD]
[/TR]
[TR]
[TD]Able to walk[/TD]
[/TR]
[TR]
[TD]Availability for duration of study period (3-4 months)[/TD]
[/TR]
[TR]
[TD]Subject using other therapies for OA, such as exercise, heat/cold therapy, joint protection and physiotherapy/occupational therapy agrees to continue these therapies as normal avoiding changes in frequency or intensity and to record therapies in the study diary[/TD]
[/TR]
[TR]
[TD]Subject agrees not to start any new therapies for OA during the course of the study[/TD]
[/TR]
[TR]
[TD]Able to give informed consent[/TD]
[/TR]
[TR]
[TD="align: center"]Exclusion Criteria[/TD]
[/TR]
[TR]
[TD]History of underlying inflammatory arthropathy; septic arthritis; inflammatory joint disease; gout; pseudogout; Paget's disease; joint fracture; acromegaly; fibromyalgia; Wilson's disease; ochronosis; haemochromatosis; heritable arthritic disorder or collagen gene mutations or rheumatoid arthritis[/TD]
[/TR]
[TR]
[TD]History of asthma, history of diabetes (Type I or Type II)[/TD]
[/TR]
[TR]
[TD]Hyperuricemia (urate, males > 480 umol/L, females > 450 umol/L)[/TD]
[/TR]
[TR]
[TD]Expectation of surgery in the next 4 months[/TD]
[/TR]
[TR]
[TD]Recent injury in the area affected by OA of the knee, i.e. meniscal tear (past 4 months)[/TD]
[/TR]
[TR]
[TD]Cartilage reconstruction procedure in the target knee[/TD]
[/TR]
[TR]
[TD]Severe OA as indicated by Lequesne's functional index score of 8 or greater, after 7 day withdrawal of usual medications[/TD]
[/TR]
[TR]
[TD]Intra-articular corticosteroid injections in the target knee within the last 3 months[/TD]
[/TR]
[TR]
[TD]Viscous injections in the target knee within the last 6 months[/TD]
[/TR]
[TR]
[TD]Hypersensitivity to NSAIDs[/TD]
[/TR]
[TR]
[TD]Abnormal liver or kidney function tests (ALT or AST > 2 times the upper limit of normal; elevated creatinine, males > 125 umol/L, females > 110 umol/L)[/TD]
[/TR]
[TR]
[TD]Abnormal findings on complete blood count[/TD]
[/TR]
[TR]
[TD]History of coagulopathies, history of peptic ulceration and upper GI hemorrhage[/TD]
[/TR]
[TR]
[TD]Uncontrolled hypertension[/TD]
[/TR]
[TR]
[TD]History of congestive heart failure, history of allergic reaction to chicken and/or eggs[/TD]
[/TR]
[TR]
[TD]History of allergic reaction to local anesthetic or to any ingredients in the test product including shellfish[/TD]
[/TR]
[TR]
[TD]Hyperkalemia (potassium > 6.2 mmol/L)[/TD]
[/TR]
[TR]
[TD]Anticipated problems with product consumption[/TD]
[/TR]
[TR]
[TD]History of cancer as well as gastrointestinal, renal, hepatic, cardiovascular, hematological, or neurological disorders[/TD]
[/TR]
[TR]
[TD]High alcohol intake (>2 standard drinks per day)[/TD]
[/TR]
[TR]
[TD]Pregnant, breastfeeding or planning to become pregnant during the study[/TD]
[/TR]
[TR]
[TD]History of psychiatric disorder that may impair the ability of subjects to provide written informed consent[/TD]
[/TR]
[TR]
[TD]Use of other natural health products, including glucosamine and chondroitin, one month prior to study and during the study, other than multivitamin and mineral supplements containing vitamins and minerals as the sole medicinal ingredients[/TD]
[/TR]
[TR]
[TD]Use of concomitant prohibited medication (narcotics, oral NSAIDs, topical NSAIDs) within four weeks of randomization[/TD]
[/TR]
[TR]
[TD]Use of acetaminophen or ibuprofen within 7 days of randomization[/TD]
[/TR]
[TR]
[TD]Subject is unwilling to stop taking pain medication other than the study medication (for arthritis or other types of pain) or is unwilling to stop taking other medications for the treatment of OA[/TD]
[/TR]
[TR]
[TD]Any other condition that, in the opinion of the investigator, would adversely affect the subject's ability to complete the study or its measures[/TD]
[/TR]
</tbody>[/TABLE]
Supplements
Each UC-II (InterHealth Nutraceuticals, Inc., Benicia, CA) capsule contained 20 mg UC-II standardized to 5 mg of bioactive undenatured type II collagen. Subjects in the UC-II group were instructed to take two “sugar pills” in the morning to protect blinding and two UC-II capsules in the evening accounting for a daily dose of 40 mg UC-II containing 10 mg of bioactive undenatured type II collagen.
Each G+C capsule contains 375 mg of glucosamine HCl (USP Grade) and 300 mg of chondroitin sulfate (USP Grade). The subjects were instructed to take two G+C capsules in the morning and two in the evening for a daily dose of 1500 mg glucosamine and 1200 mg chondroitin.
Removal of Patients from Therapy or Assessment
The criteria for removal of patients from the study included:
Adverse events
For any adverse event, patients were examined and appropriately managed or the patients would be referred to another medical professional for proper evaluation and treatment. If medical problems were attributed to the trial compounds, then the trial drugs were discontinued and the toxicities were reported.
Personal reasons
As stated in the Consent Form, subjects were able to withdraw from the study for any reason at any time.
Clinical judgment of physician
Subjects were withdrawn from the study (without penalty) if, in the opinion of the treating physician, it was not in the patient's best interest to continue. For instance, if during the course of the study a patient became pregnant, she would be withdrawn from the study because it was not known how the study compounds/medications might affect an unborn child.
Protocol violation
Any subject found to have entered this study in violation of the protocol or failed to follow the study protocol were discontinued from the study at the discretion of the Principal Investigator. Subjects were withdrawn for protocol non-compliance if they adhered to the dosing schedule less than 75% of the time.
Method of assigning patients to treatment groups
Patients were assigned to treatment groups (order of treatments) using computer-generated randomization tables. Patients were not stratified or assigned using any other specific method and were not randomized after stratification or blocking procedures.
Selection of doses in the study
The justification for the daily dose of 40 mg UC-II in capsules (providing 10 mg of undenatured collagen II) is based on efficacy demonstrated in earlier studies (8,9).
Blinding
In order to protect blinding, subjects were given bottles containing product labeled with “AM” or “PM” to distinguish the time in which treatment was to be taken. Each bottle contained descriptions of all potential products to ensure blinding was protected. Additionally, each bottle was labeled with a randomization number. In the event that an adverse effect was considered serious and related to the investigational product, the blind would be broken for that individual subject.
Neither the patient, nor investigator, nor research staff, were aware which test compound the subject was assigned. Interim analysis was performed in order to write a preliminary report and thus preliminary unblinding occurred by an individual unrelated to the study conduct. Personnel related to analysis, statistics, and report writing remained blinded.
Prior and concomitant therapy
Uses of medications such as narcotics, oral NSAIDS, topical NSAIDS within four weeks of randomization and during the study, were not allowed.
Treatment compliance
Compliance was assessed by capsule count at visits 3, 4, and 5 and review of subject diary.
Efficacy and Safety Variables
Efficacy and safety measurements assessed
Adverse events
During the study, subjects recorded adverse effects in their subject diary. At each visit, the subjects were asked if they experienced problems or difficulties. Any adverse events were documented and recorded in the study record and was classified according to the description, duration, severity, frequency, and outcome. The investigator assessed the adverse events and decided causality. Classifications were as per the Coding Symbol Thesaurus of Adverse Reaction Terms (COSTART) U.S. Food and Drug Administration (16).
Blood tests
Blood samples were taken from all subjects during screening (visit 1) and at end of study (visit 5). Blood samples (approximately 15 ml) were taken from subjects at day 30 and day 60 (visits 3 and 4) for the determination of ALT, AST, bilirubin, and albumin if the subjects had been taking acetaminophen greater than 2 g/day for more than 7 days. All blood samples were analyzed by MDS Laboratory Services (London, Ontario, Canada).
Appropriateness of Measurements
The efficacy and safety assessments used in this study were standard for OA and are widely used and recognized as reliable, accurate, and relevant. WOMAC scores were determined, at screening, and baseline, as well as at days 30, 60 and 90 as described in Bellamy et al (17). Other objectives also performed at days 0, 30, 60 and 90 included determination of Lequesne's functional index, VAS pain scores, knee flexion, time to walk 50 m, time to climb 10 steps, physician's and subject's global assessment. The Lequesne's functional index is described in Lequesne et al. (18).
Statistical Methods
Sample size of 25 subjects per group was based on the subject number used in Braham et al. (1). To compare UC-II with G+C group, a linear contrast was included in the analysis of variance. Data missing subsequent to 30 days were imputed using the last-observation-carried forward technique. Furthermore, comparisons between the UC-II and G+C groups were made at each visit using analysis of variance, using the baseline visit as a covariate. SAS version 9.1 has been used to perform the statistical analysis. Probability values less than 0.05 were considered statistically significant for between-group comparisons.
Results
Baseline Statistics and Compliance of Trial Subjects
Demographic and baseline characteristics of patients are summarized in Table 3. Overall, the patient profiles with respect to age, sex, height, weight, blood pressure, heart beat and target knee were similar between both treatment groups. Table 4 shows treatment compliance of the trial patients. There were no significant interaction terms or between-group differences for compliances. When compliances were compared at each visit, there were no overall between-group differences among the two treatment groups.
Table 3 Demographic and baseline characteristics of the trial subjects
[TABLE="class: ivytablelight"]
<tbody>[TR]
[TH="colspan: 2"][/TH]
[TH]UC-II (N=26)[/TH]
[TH]G + C (N=26)[/TH]
[/TR]
[TR]
[TD="colspan: 2"]Age (years)[/TD]
[TD]58.9 ± 9.79[/TD]
[TD]58.7 ± 10.3[/TD]
[/TR]
[TR]
[TD="colspan: 2"]Sex: male/female (%)[/TD]
[TD]13/26 (50%)[/TD]
[TD]17/26 (65%)[/TD]
[/TR]
[TR]
[TD="colspan: 2"]Height (cm)[/TD]
[TD]167.7 ± 9.90[/TD]
[TD]167.0 ± 8.73[/TD]
[/TR]
[TR]
[TD="colspan: 2"]Weight (kg)[/TD]
[TD]84.3 ± 17.4[/TD]
[TD]86.6 ± 21.0[/TD]
[/TR]
[TR]
[TD="colspan: 2"]Systolic Blood Pressure (mm)[/TD]
[TD]128.2 ± 9.36[/TD]
[TD]126.3 ± 12.5[/TD]
[/TR]
[TR]
[TD="colspan: 2"]Diastolic Blood Pressure (mm)[/TD]
[TD]81.9 ± 7.43[/TD]
[TD]79.7 ± 8.60[/TD]
[/TR]
[TR]
[TD="colspan: 2"]Heart Rate (bpm)[/TD]
[TD]68.2 ± 7.72[/TD]
[TD]67.4 ± 8.47[/TD]
[/TR]
[TR]
[TD="colspan: 2"]Target knee[/TD]
[TD][/TD]
[TD][/TD]
[/TR]
[TR]
[TD][/TD]
[TD]Left; n (%)[/TD]
[TD]16 (61.5%)[/TD]
[TD]13 (50%)[/TD]
[/TR]
[TR]
[TD]Right; n (%)[/TD]
[TD]10 (38.5%)[/TD]
[TD]13 (50%)[/TD]
[/TR]
[TR]
[TD="colspan: 4"]Where applicable, values are expressed as mean ± SD[/TD]
[/TR]
</tbody>[/TABLE]
Table 4 Treatment compliance as assessed during specified visits
[TABLE="class: ivytablelight"]
<tbody>[TR]
[TH]Visit[/TH]
[TH="colspan: 2"]Treatment Group[/TH]
[/TR]
[TR]
[TH]UC-II[/TH]
[TH]G + C[/TH]
[/TR]
[TR]
[TD="colspan: 3"]AM Capsule Compliance[/TD]
[/TR]
[TR]
[TD]Visit 3[/TD]
[TD][25] 90.5 ± 19.2[/TD]
[TD][25] 93.6 ± 11.5[/TD]
[/TR]
[TR]
[TD]Visit 4[/TD]
[TD][24] 93.2 ± 9.66[/TD]
[TD][26] 94.5 ± 11.8[/TD]
[/TR]
[TR]
[TD]Visit 5[/TD]
[TD][23] 98.5 ± 5.15[/TD]
[TD][26] 93.3 ± 11.0[/TD]
[/TR]
[TR]
[TD="colspan: 3"]PM Capsule Compliance[/TD]
[/TR]
[TR]
[TD]Visit 3[/TD]
[TD][25] 88.1 ± 18.7[/TD]
[TD][25] 92.5 ± 12.5[/TD]
[/TR]
[TR]
[TD]Visit 4[/TD]
[TD][24] 92.8 ± 8.97[/TD]
[TD][26] 91.6 ± 12.3[/TD]
[/TR]
[TR]
[TD]Visit 5[/TD]
[TD][22] 95.3 ± 9.92[/TD]
[TD][26] 89.7 ± 12.6[/TD]
[/TR]
</tbody>[/TABLE]
There were no significant interaction terms and between-group differences for compliances. When compliances were compared at each visit, there were no overall between-group differences among the five treatment groups. Values are expressed as [n] mean ± SD.
WOMAC Score
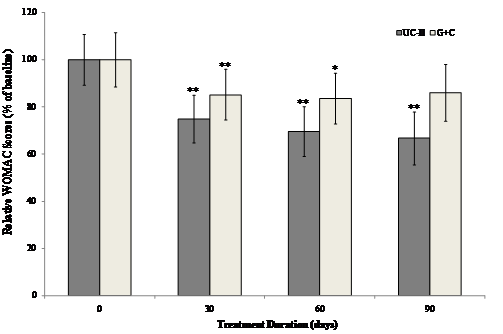
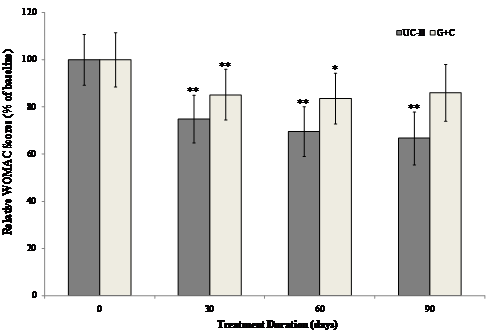
The interaction between visit and treatment was significant in UC-II treated group for "pain walking on flat surface" (p=0.034), "difficulty walking on flat surface" (p=0.038) and "performing heavy domestic duties" (p=0.031) as compared to G+C treated group. There was evidence that UC-II treatment has a significant effect for “ascending stairs” (p=0.013) as compared to G+C treatment. Additionally, when groups were compared at each visit, UC-II was significantly better than G+C for “ascending stairs at 30 days and 60 days” (p=0.019 & 0.040 respectively), “at night while in bed” (p=0.015) at 60 days and difficulty walking on flat surface at 90 days (p=0.035). There were no further statistically significant differences for any other individual WOMAC components or summary scores. Treatment with UC-II was most effective and reduced the WOMAC scores by 33% compared to 14% in (G+C)-treated groups after 90 days. Within-group analysis indicated that treatment with UC-II for 90 days significantly (p<0.05) improved WOMAC scores at all treatment time points measured. In contrast, subjects received G+C did not show any statistical significant change in WOMAC scores at Day 90 of treatment (Fig. 2).
VAS Score
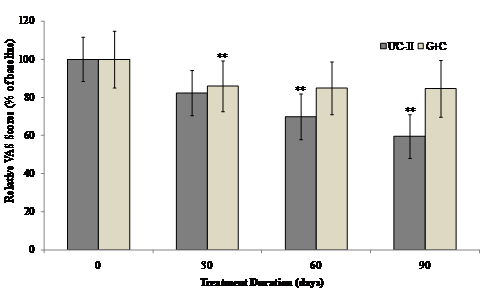
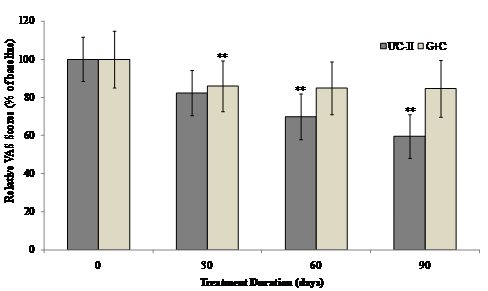
The interaction between visit and treatment was non-significant for all VAS components and summary scores. However there was evidence that UC-II treatment had a significant effect for “pain during climbing up and down stairs”, “night pain” and “resting pain” (p=0.035, 0.030 and 0.024 respectively). When groups were compared at each visit, UC-II was significantly better than G+C for “night pain” (p=0.040) and “resting pain” (p=0.020) at 60 days and “pain during climbing up and down stairs” (p=0.014) and “resting pain” at 90 days (p=0.034). There were no between-group differences for any of the VAS components or summary scores. Although both the treatments reduced the VAS score, UC-II was found to be more effective with a 40% decrease after 90 days of treatment compared to a 15% decrease in G+C treated groups.
Within-group analysis indicated that subjects on UC-II showed a significant reduction in total VAS scores at Day 60 and Day 90 as compared to baseline. However, subjects on G+C showed a significant reduction in total VAS scores at Day 30 and no significant difference was observed at either Day 60 or Day 90 as compared to baseline (Fig. 3).
Figure 2 Changes in WOMAC scores at Day 90 from baseline. WOMAC scores from each treatment group were compared to baseline value at specified time points. Each bar presents mean ± SEM. *p<0.05, **p<0.005 indicate significantly different from baseline.
 (Click on the image to enlarge.)
(Click on the image to enlarge.)
Figure 3 Changes in VAS score at Day 90 from baseline. VAS scores from each treatment group were compared to baseline value at specified time points. Each bar presents mean ± SEM. **p<0.05 indicates significantly different from baseline.
 (Click on the image to enlarge.)
(Click on the image to enlarge.)
Lequesne Score
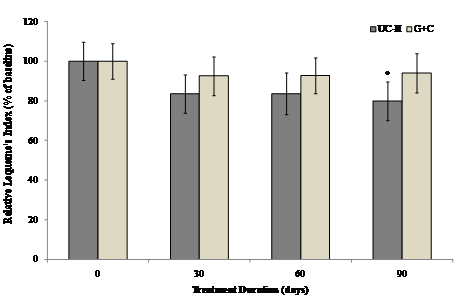
The Lequesne's functional index was used to determine the effect of different treatments on pain during daily activities. The interaction between visit and treatment was non-significant for all Lequesne's components and summary scores. Furthermore, there were no between-group differences for any of the Lequesne's components or summary scores. However there was evidence that visit has a significant effect in UC-II treated group for “pain while up from sitting” and “maximum distance walked” (p=0.036 and 0.002 respectively) as compared to G+C treated group. There was as a strong trend toward UC-II efficacy. UC-II treatment effectively reduced Lequesne's functional index score by 20.1% as compared to 5.9 % by G+C treatment.
Within-group analysis suggested that subjects on UC-II demonstrated a significant reduction in total Lequesne's index of severity score from baseline to Day 90, whereas no significant difference from baseline was observed for subjects on G+C at any treatment time points evaluated (Fig. 4).
Figure 4 Changes in Lequesne's functional index at Day 90 from baseline. Lequesne's functional index from each treatment group was compared to baseline value at specified time points. Each bar presents mean ± SEM. *p<0.05 indicates significantly different from baseline.
 (Click on the image to enlarge.)
(Click on the image to enlarge.)
Adverse Events
Adverse effects that occurred during the 90-day trial period are summarized in Table 5. Overall, there were 58 adverse events noted in the subjects receiving G+C treatment, whereas, only 35 adverse events were observed in UC-II group. In terms of severity, 60% of mild and 38% of moderate adverse events were experienced by subjects on G+C in comparison to 43% and 54% by subjects on UC-II. In relationship to test product a higher number of subjects (23%) on G+C demonstrated adverse events possibly related to product as compared to 11.4% of subjects on UC-II. For UC-II the possible adverse events related to products were constipation and headaches (intermittently). For G+C the possible adverse events related to products were bloating, stomach pain, rash, water retention (edema around eyes and scars), hives on face and chest, and headache. However, there was no significant difference in the occurrence of adverse effects between the two treatment groups.
Rescue Medication
A greater percentage of subjects used rescue medication while on G+C as compared to UC-II at every time point assessed. From baseline to Day 30 a total of 8 subjects (33.3%) on UC-II used rescue medication as compared to 23 subjects (88.5%) on G+C. From Day 30 to Day 60, 13 subjects (54.2%) on UC-II used rescue medication as compared to 21 subjects (80.8%) on G+C. Fourteen subjects (63.6%) on UC-II used rescue medication as compared to 19 subjects (79.2%) on G+C from Day 60 to Day 90.
Table 5 Summary of analysis of adverse events in all subjects
[TABLE="class: ivytablelight"]
<tbody>[TR]
[TH][/TH]
[TH="colspan: 2"]Treatment Group[/TH]
[/TR]
[TR]
[TH]UC-II (n=26)[/TH]
[TH]G + C (n=26)[/TH]
[/TR]
[TR]
[TD="colspan: 3"]Severity") [/TD]
[/TD]
[/TR]
[TR]
[TD]Mild[/TD]
[TD]15[/TD]
[TD]35[/TD]
[/TR]
[TR]
[TD]Moderate[/TD]
[TD]19[/TD]
[TD]22[/TD]
[/TR]
[TR]
[TD]Severe[/TD]
[TD]1[/TD]
[TD]1[/TD]
[/TR]
[TR]
[TD="colspan: 3"]Relationship to Test Article[/TD]
[/TR]
[TR]
[TD]Not related[/TD]
[TD]17[/TD]
[TD]20[/TD]
[/TR]
[TR]
[TD]Unlikely[/TD]
[TD]14[/TD]
[TD]30[/TD]
[/TR]
[TR]
[TD]Possible[/TD]
[TD]4[/TD]
[TD]8[/TD]
[/TR]
[TR]
[TD]Probable[/TD]
[TD]0[/TD]
[TD]0[/TD]
[/TR]
[TR]
[TD]Most Probable[/TD]
[TD]0[/TD]
[TD]0[/TD]
[/TR]
[TR]
[TD="colspan: 3"]Body System[/TD]
[/TR]
[TR]
[TD]Pain[/TD]
[TD]10[/TD]
[TD]17[/TD]
[/TR]
[TR]
[TD]Gastrointestinal[/TD]
[TD]5[/TD]
[TD]15[/TD]
[/TR]
[TR]
[TD]Musculoskeletal/Soft Tissue[/TD]
[TD]7[/TD]
[TD]5[/TD]
[/TR]
[TR]
[TD]Neurology[/TD]
[TD]0[/TD]
[TD]2[/TD]
[/TR]
[TR]
[TD]Pulmonary / Upper Respiratory[/TD]
[TD]2[/TD]
[TD]1[/TD]
[/TR]
[TR]
[TD]Hemorrhage/Bleeding[/TD]
[TD]2[/TD]
[TD]1[/TD]
[/TR]
[TR]
[TD]Blood/Bone Marrow[/TD]
[TD]2[/TD]
[TD]1[/TD]
[/TR]
[TR]
[TD]Dermatology/Skin[/TD]
[TD]2[/TD]
[TD]3[/TD]
[/TR]
[TR]
[TD]Allergy / Immunology[/TD]
[TD]0[/TD]
[TD]1[/TD]
[/TR]
[TR]
[TD]Infection[/TD]
[TD]1[/TD]
[TD]3[/TD]
[/TR]
[TR]
[TD]Lymphatics[/TD]
[TD]0[/TD]
[TD]1[/TD]
[/TR]
[TR]
[TD]Hepatobilary / Pancreatic[/TD]
[TD]0[/TD]
[TD]0[/TD]
[/TR]
[TR]
[TD]Renal / Genitoruinary[/TD]
[TD]0[/TD]
[TD]0[/TD]
[/TR]
[TR]
[TD]Constitutional Symptoms[/TD]
[TD]2[/TD]
[TD]3[/TD]
[/TR]
[TR]
[TD]Syndromes[/TD]
[TD]1[/TD]
[TD]1[/TD]
[/TR]
[TR]
[TD]Auditory/Ear[/TD]
[TD]0[/TD]
[TD]1[/TD]
[/TR]
[TR]
[TD]Ocular / Visual[/TD]
[TD]0[/TD]
[TD]1[/TD]
[/TR]
[TR]
[TD]Metabolic / Laboratory[/TD]
[TD]1[/TD]
[TD]2[/TD]
[/TR]
[TR]
[TD]Total Number of Adverse Events Experienced During Study[/TD]
[TD]35[/TD]
[TD]58[/TD]
[/TR]
[TR]
[TD]Total Number of Subjects Experiencing Adverse Events: n (%)[/TD]
[TD]16/26 (61.5%)[/TD]
[TD]20/26 (76.9%)[/TD]
[/TR]
</tbody>[/TABLE]
see part 2
Int J Med Sci 2009; 6(6):312-321. doi:10.7150/ijms.6.312
Research Paper
Safety and efficacy of undenatured type II collagen in the treatment of osteoarthritis of the knee: a clinical trial
David C. Crowley[SUP]1[/SUP], Francis C. Lau[SUP]2[/SUP], Prachi Sharma[SUP]1[/SUP], Malkanthi Evans[SUP]1[/SUP], Najla Guthrie[SUP]1[/SUP], Manashi Bagchi[SUP]2[/SUP], Debasis Bagchi[SUP]2,3[/SUP], Dipak K. Dey[SUP]4[/SUP], Siba P. Raychaudhuri[SUP] 5,6,
[/SUP]1. KGK Synergize Incorporated, London, ON, Canada
2. Department of Research and Development, InterHealth Research Center, Benicia, CA, USA
3. Department of Pharmacology and Pharmaceutical Sciences, University of Houston College of Pharmacy, Houston, TX, USA
4. Department of Statistics, University of Connecticut, Storrs, CT, USA
5. Department of Medicine, Division of Rheumatology, Allergy and Immunology, School of Medicine, University of California Davis, Davis, CA, USA
6. VA Medical Center Sacramento, Hospital Way, Mather, CA, USA
How to cite this article:
Crowley DC, Lau FC, Sharma P, Evans M, Guthrie N, Bagchi M, Bagchi D, Dey DK, Raychaudhuri SP. Safety and efficacy of undenatured type II collagen in the treatment of osteoarthritis of the knee: a clinical trial. Int J Med Sci 2009; 6(6):312-321. doi:10.7150/ijms.6.312. Available from http://www.medsci.org/v06p0312.htm
Abstract
Previous studies have shown that undenatured type II collagen (UC-II) is effective in the treatment of rheumatoid arthritis, and preliminary human and animal trials have shown it to be effective in treating osteoarthritis (OA). The present clinical trial evaluated the safety and efficacy of UC-II as compared to a combination of glucosamine and chondroitin (G+C) in the treatment of OA of the knee. The results indicate that UC-II treatment was more efficacious resulting in a significant reduction in all assessments from the baseline at 90 days; whereas, this effect was not observed in G+C treatment group. Specifically, although both treatments reduced the Western Ontario McMaster Osteoarthritis Index (WOMAC) score, treatment with UC-II reduced the WOMAC score by 33% as compared to 14% in G+C treated group after 90 days. Similar results were obtained for visual analog scale (VAS) scores. Although both the treatments reduced the VAS score, UC-II treatment decreased VAS score by 40% after 90 days as compared to 15.4% in G+C treated group. The Lequesne's functional index was used to determine the effect of different treatments on pain during daily activities. Treatment with UC-II reduced Lequesne's functional index score by 20% as compared to 6% in G+C treated group at the end of 90-day treatment. Thus, UC-II treated subjects showed significant enhancement in daily activities suggesting an improvement in their quality of life.
Keywords: undenatured type II collagen, osteoarthritis, glucosamine, chondroitin, WOMAC, visual analog scale, Lequesne's Functional Index
INTRODUCTION
Arthritis afflicts approximately 43 million Americans or approximately 16.6% of the US population. The two most common types of arthritis are osteoarthritis (OA) and rheumatoid arthritis (RA). OA of the knee and hip is a growing health concern and is the most common forms of arthritis (1-3). Pain and disease can range from very mild to very severe (3). Patients with OA have pain that typically worsens with weight bearing, including walking and standing, and improves with rest (4). Other symptoms include morning stiffness and gelling of the involved joint after periods of inactivity. Currently, OA affects nearly 21 million people in the United States, accounting for 25% of visits to primary care physicians, and half of all Non-Steroidal Anti-Inflammatory Drugs (NSAID) prescriptions. The diverse clinical patterns of OA are observed in approximately 10% of people older than 60 years thus compromising the quality of life of millions of Americans. In addition, OA costs the North American economy approximately $60 billion per year.
Current treatment of OA includes exercise, heat/cold therapy, joint protection, weight loss, physiotherapy/occupational therapy and medications (3-5). The most common medications include acetaminophen and NSAIDs. Although these drugs are effective for reducing pain associated with OA, they do not reverse the disease. In addition, there are considerable side effects associated with the use of these drugs. As a result, OA sufferers have turned to natural nutraceuticals to ease their pain and discomfort. These products are commonly used because they are well tolerated and considered safe. Nutraceuticals are defined as functional foods, natural products, or parts of food that provide medicinal, therapeutic, or health benefits, including the prevention or treatment of disease. Currently, glucosamine and chondroitin are the two most commonly used nutraceuticals in humans as well as in animals to alleviate pain associated with arthritis (6). However, recent randomized controlled trials and meta-analysis of these supplements have shown only small-to-moderate symptomatic efficacy in human OA (7). An emerging novel nutraceutical ingredient known as UC-II has received considerable attention in the treatment of OA. UC-II is a novel undenatured type II collagen derived from chicken sternum cartilage. Previous studies have shown that undenatured type II collagen is effective in the treatment of RA (8-11), and preliminary human (12) and animal (13) trials have shown it to be effective in treating OA. Obese-arthritic dogs given 4 mg or 40 mg daily dose of UC-II for 90 days showed significant declines in overall pain, pain during limb manipulation and lameness after physical exertion (14). Greater improvement was observed with the 40 mg dose. No adverse effects or significant changes in serum chemistry were noted. Following UC-II withdrawal for a period of 30 days, all dogs experienced a relapse of overall pain, exercise-associated lameness and pain upon limb manipulation. Studies have also shown that small doses of orally administered undenatured type II chicken collagen inhibit killer T-cell attack (15). The present clinical trial evaluated the safety and efficacy of UC-II in the treatment of the knee in OA patients.
Materials and Methods
Study Design
This clinical trial (Human Clinical Trial Approval #06UOHI) was managed by KGK Synergize Inc. (London, ON, Canada). The study was conducted at two sites: 1) KGK Synergize Inc., and 2) Corunna Medical Research (Corunna, ON, Canada). Figure 1 illustrates the study design while Table 1 lists the procedures and observations at each time point. Briefly, at screening (Visit 1) the consent form was discussed, signed and a complete physical examination was performed. Activity level, diet history, medication/supplement use and medical history were recorded. The VAS score, the WOMAC Index and Lequesne scores were obtained. Urine was collected for a pregnancy test for women of childbearing potential. A blood sample was taken for determination of uric acid, CBC count and differentiation, albumin, total protein, sodium, potassium, chloride, BUN, creatinine, ALT, AST, bilirubin, erythrocyte sedimentation rate (ESR) and rheumatoid factor. Upon review of blood test results, eligible subjects were instructed to get an X-ray of the affected knees to confirm diagnosis. A total of 52 subjects were recruited using the inclusion and exclusion criteria outlined in Table 2. At the first treatment visit (Visit 2), selected subjects were randomly assigned to receive UC-II (n = 26) or glucosamine HCl plus chondroitin sulfate (n = 26, G+C). On each test day (day 0, 30, 60, 90), subjects were required to come to the clinic for clinical assessment. The clinical assessments included WOMAC, Lequesne's functional index and 100-mm VAS pain scores. A subject treatment diary was completed by each patient throughout the study period to determine side effects, medication use, and product compliance.
Figure 1 UC-II clinical study design. The study was a two-site, randomized, double-blind study conducted in London, Ontario and Corunna, Ontario, Canada.
 (Click on the image to enlarge.)
(Click on the image to enlarge.)Table 1 Schedule of observations and procedures
[TABLE="class: ivytablelight"]
<tbody>[TR]
[TH]Procedure[/TH]
[TH]Visit 1 Screening[/TH]
[TH]Visit 2 Day 0[/TH]
[TH]Visit 3 Day 30[/TH]
[TH]Visit 4 Day 60[/TH]
[TH]Visit 5 Day 90[/TH]
[/TR]
[TR]
[TD]Informed consent[/TD]
[TD]X[/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[/TR]
[TR]
[TD]Review inclusion/exclusion[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[/TR]
[TR]
[TD]Medical history including activity level and diet history[/TD]
[TD]X[/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[/TR]
[TR]
[TD]Physical examination[/TD]
[TD]X[/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[/TR]
[TR]
[TD]Biometric measurements: Weight, height*, heart rate and blood pressure.[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[/TR]
[TR]
[TD]Urine pregnancy test[/TD]
[TD]X[/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[/TR]
[TR]
[TD]Concomitant medications[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[/TR]
[TR]
[TD]Blood samples: Uric acid, CBC count and differentiation, albumin, total protein, sodium, potassium, chloride, BUN, creatinine, ALT, AST, bilirubin, ESR, rheumatoid factor[/TD]
[TD]X[/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[TD]X[/TD]
[/TR]
[TR]
[TD]WOMAC, VAS and Lequesne scores[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[/TR]
[TR]
[TD]X-ray[/TD]
[TD]X[/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[/TR]
[TR]
[TD]Randomization[/TD]
[TD][/TD]
[TD]X[/TD]
[TD][/TD]
[TD][/TD]
[TD][/TD]
[/TR]
[TR]
[TD]Blood sample: ALT, AST, bilirubin, albumin.[/TD]
[TD][/TD]
[TD][/TD]
[TD]X[SUP]†[/SUP][/TD]
[TD]X[SUP]†[/SUP][/TD]
[TD][/TD]
[/TR]
[TR]
[TD]Knee flexion, Time to walk 50m, Swelling in the knee joint, Time for climbing 10 steps[/TD]
[TD][/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[/TR]
[TR]
[TD]Physician's Global Assessment[/TD]
[TD][/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[/TR]
[TR]
[TD]Subject's Global Assessment[/TD]
[TD][/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[/TR]
[TR]
[TD]Investigational Product dispensed[/TD]
[TD][/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD][/TD]
[/TR]
[TR]
[TD]Subject Treatment Diary dispensed[/TD]
[TD][/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD][/TD]
[/TR]
[TR]
[TD]Investigational Product returned Compliance calculated[/TD]
[TD][/TD]
[TD][/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[/TR]
[TR]
[TD]Subject Treatment Diary returned[/TD]
[TD][/TD]
[TD][/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[/TR]
[TR]
[TD]Adverse Events[/TD]
[TD][/TD]
[TD][/TD]
[TD]X[/TD]
[TD]X[/TD]
[TD]X[/TD]
[/TR]
</tbody>[/TABLE]
* height was only measured at visit 1
[SUP]† [/SUP]If acetaminophen use was greater than 2 g/day for more than 7 days
Table 2 Inclusion and exclusion criteria
[TABLE="class: ivytablelight"]
<tbody>[TR]
[TH="align: center"]Inclusion Criteria[/TH]
[/TR]
[TR]
[TD]Males and females 40-75 years old[/TD]
[/TR]
[TR]
[TD]Females of childbearing potential must agree to use a medically approved form of birth control and have a negative urine pregnancy test result[/TD]
[/TR]
[TR]
[TD]Unilateral or bilateral OA of the knee for greater than 3 months (American College of Rheumatology criteria) confirmed by radiologist's report, i.e. X-rays showing osteophytes, joint space narrowing or subchondral bone sclerosis (eburnation)[/TD]
[/TR]
[TR]
[TD]Erythrocyte sedimentation rate (ESR) < 40 mm/hr[/TD]
[/TR]
[TR]
[TD]Moderate OA as indicated by Lequesne's functional index score of 4.5-7.5 after 7 day withdrawal of usual medications[/TD]
[/TR]
[TR]
[TD]Able to walk[/TD]
[/TR]
[TR]
[TD]Availability for duration of study period (3-4 months)[/TD]
[/TR]
[TR]
[TD]Subject using other therapies for OA, such as exercise, heat/cold therapy, joint protection and physiotherapy/occupational therapy agrees to continue these therapies as normal avoiding changes in frequency or intensity and to record therapies in the study diary[/TD]
[/TR]
[TR]
[TD]Subject agrees not to start any new therapies for OA during the course of the study[/TD]
[/TR]
[TR]
[TD]Able to give informed consent[/TD]
[/TR]
[TR]
[TD="align: center"]Exclusion Criteria[/TD]
[/TR]
[TR]
[TD]History of underlying inflammatory arthropathy; septic arthritis; inflammatory joint disease; gout; pseudogout; Paget's disease; joint fracture; acromegaly; fibromyalgia; Wilson's disease; ochronosis; haemochromatosis; heritable arthritic disorder or collagen gene mutations or rheumatoid arthritis[/TD]
[/TR]
[TR]
[TD]History of asthma, history of diabetes (Type I or Type II)[/TD]
[/TR]
[TR]
[TD]Hyperuricemia (urate, males > 480 umol/L, females > 450 umol/L)[/TD]
[/TR]
[TR]
[TD]Expectation of surgery in the next 4 months[/TD]
[/TR]
[TR]
[TD]Recent injury in the area affected by OA of the knee, i.e. meniscal tear (past 4 months)[/TD]
[/TR]
[TR]
[TD]Cartilage reconstruction procedure in the target knee[/TD]
[/TR]
[TR]
[TD]Severe OA as indicated by Lequesne's functional index score of 8 or greater, after 7 day withdrawal of usual medications[/TD]
[/TR]
[TR]
[TD]Intra-articular corticosteroid injections in the target knee within the last 3 months[/TD]
[/TR]
[TR]
[TD]Viscous injections in the target knee within the last 6 months[/TD]
[/TR]
[TR]
[TD]Hypersensitivity to NSAIDs[/TD]
[/TR]
[TR]
[TD]Abnormal liver or kidney function tests (ALT or AST > 2 times the upper limit of normal; elevated creatinine, males > 125 umol/L, females > 110 umol/L)[/TD]
[/TR]
[TR]
[TD]Abnormal findings on complete blood count[/TD]
[/TR]
[TR]
[TD]History of coagulopathies, history of peptic ulceration and upper GI hemorrhage[/TD]
[/TR]
[TR]
[TD]Uncontrolled hypertension[/TD]
[/TR]
[TR]
[TD]History of congestive heart failure, history of allergic reaction to chicken and/or eggs[/TD]
[/TR]
[TR]
[TD]History of allergic reaction to local anesthetic or to any ingredients in the test product including shellfish[/TD]
[/TR]
[TR]
[TD]Hyperkalemia (potassium > 6.2 mmol/L)[/TD]
[/TR]
[TR]
[TD]Anticipated problems with product consumption[/TD]
[/TR]
[TR]
[TD]History of cancer as well as gastrointestinal, renal, hepatic, cardiovascular, hematological, or neurological disorders[/TD]
[/TR]
[TR]
[TD]High alcohol intake (>2 standard drinks per day)[/TD]
[/TR]
[TR]
[TD]Pregnant, breastfeeding or planning to become pregnant during the study[/TD]
[/TR]
[TR]
[TD]History of psychiatric disorder that may impair the ability of subjects to provide written informed consent[/TD]
[/TR]
[TR]
[TD]Use of other natural health products, including glucosamine and chondroitin, one month prior to study and during the study, other than multivitamin and mineral supplements containing vitamins and minerals as the sole medicinal ingredients[/TD]
[/TR]
[TR]
[TD]Use of concomitant prohibited medication (narcotics, oral NSAIDs, topical NSAIDs) within four weeks of randomization[/TD]
[/TR]
[TR]
[TD]Use of acetaminophen or ibuprofen within 7 days of randomization[/TD]
[/TR]
[TR]
[TD]Subject is unwilling to stop taking pain medication other than the study medication (for arthritis or other types of pain) or is unwilling to stop taking other medications for the treatment of OA[/TD]
[/TR]
[TR]
[TD]Any other condition that, in the opinion of the investigator, would adversely affect the subject's ability to complete the study or its measures[/TD]
[/TR]
</tbody>[/TABLE]
Supplements
Each UC-II (InterHealth Nutraceuticals, Inc., Benicia, CA) capsule contained 20 mg UC-II standardized to 5 mg of bioactive undenatured type II collagen. Subjects in the UC-II group were instructed to take two “sugar pills” in the morning to protect blinding and two UC-II capsules in the evening accounting for a daily dose of 40 mg UC-II containing 10 mg of bioactive undenatured type II collagen.
Each G+C capsule contains 375 mg of glucosamine HCl (USP Grade) and 300 mg of chondroitin sulfate (USP Grade). The subjects were instructed to take two G+C capsules in the morning and two in the evening for a daily dose of 1500 mg glucosamine and 1200 mg chondroitin.
Removal of Patients from Therapy or Assessment
The criteria for removal of patients from the study included:
Adverse events
For any adverse event, patients were examined and appropriately managed or the patients would be referred to another medical professional for proper evaluation and treatment. If medical problems were attributed to the trial compounds, then the trial drugs were discontinued and the toxicities were reported.
Personal reasons
As stated in the Consent Form, subjects were able to withdraw from the study for any reason at any time.
Clinical judgment of physician
Subjects were withdrawn from the study (without penalty) if, in the opinion of the treating physician, it was not in the patient's best interest to continue. For instance, if during the course of the study a patient became pregnant, she would be withdrawn from the study because it was not known how the study compounds/medications might affect an unborn child.
Protocol violation
Any subject found to have entered this study in violation of the protocol or failed to follow the study protocol were discontinued from the study at the discretion of the Principal Investigator. Subjects were withdrawn for protocol non-compliance if they adhered to the dosing schedule less than 75% of the time.
Method of assigning patients to treatment groups
Patients were assigned to treatment groups (order of treatments) using computer-generated randomization tables. Patients were not stratified or assigned using any other specific method and were not randomized after stratification or blocking procedures.
Selection of doses in the study
The justification for the daily dose of 40 mg UC-II in capsules (providing 10 mg of undenatured collagen II) is based on efficacy demonstrated in earlier studies (8,9).
Blinding
In order to protect blinding, subjects were given bottles containing product labeled with “AM” or “PM” to distinguish the time in which treatment was to be taken. Each bottle contained descriptions of all potential products to ensure blinding was protected. Additionally, each bottle was labeled with a randomization number. In the event that an adverse effect was considered serious and related to the investigational product, the blind would be broken for that individual subject.
Neither the patient, nor investigator, nor research staff, were aware which test compound the subject was assigned. Interim analysis was performed in order to write a preliminary report and thus preliminary unblinding occurred by an individual unrelated to the study conduct. Personnel related to analysis, statistics, and report writing remained blinded.
Prior and concomitant therapy
Uses of medications such as narcotics, oral NSAIDS, topical NSAIDS within four weeks of randomization and during the study, were not allowed.
Treatment compliance
Compliance was assessed by capsule count at visits 3, 4, and 5 and review of subject diary.
Efficacy and Safety Variables
Efficacy and safety measurements assessed
Adverse events
During the study, subjects recorded adverse effects in their subject diary. At each visit, the subjects were asked if they experienced problems or difficulties. Any adverse events were documented and recorded in the study record and was classified according to the description, duration, severity, frequency, and outcome. The investigator assessed the adverse events and decided causality. Classifications were as per the Coding Symbol Thesaurus of Adverse Reaction Terms (COSTART) U.S. Food and Drug Administration (16).
Blood tests
Blood samples were taken from all subjects during screening (visit 1) and at end of study (visit 5). Blood samples (approximately 15 ml) were taken from subjects at day 30 and day 60 (visits 3 and 4) for the determination of ALT, AST, bilirubin, and albumin if the subjects had been taking acetaminophen greater than 2 g/day for more than 7 days. All blood samples were analyzed by MDS Laboratory Services (London, Ontario, Canada).
Appropriateness of Measurements
The efficacy and safety assessments used in this study were standard for OA and are widely used and recognized as reliable, accurate, and relevant. WOMAC scores were determined, at screening, and baseline, as well as at days 30, 60 and 90 as described in Bellamy et al (17). Other objectives also performed at days 0, 30, 60 and 90 included determination of Lequesne's functional index, VAS pain scores, knee flexion, time to walk 50 m, time to climb 10 steps, physician's and subject's global assessment. The Lequesne's functional index is described in Lequesne et al. (18).
Statistical Methods
Sample size of 25 subjects per group was based on the subject number used in Braham et al. (1). To compare UC-II with G+C group, a linear contrast was included in the analysis of variance. Data missing subsequent to 30 days were imputed using the last-observation-carried forward technique. Furthermore, comparisons between the UC-II and G+C groups were made at each visit using analysis of variance, using the baseline visit as a covariate. SAS version 9.1 has been used to perform the statistical analysis. Probability values less than 0.05 were considered statistically significant for between-group comparisons.
Results
Baseline Statistics and Compliance of Trial Subjects
Demographic and baseline characteristics of patients are summarized in Table 3. Overall, the patient profiles with respect to age, sex, height, weight, blood pressure, heart beat and target knee were similar between both treatment groups. Table 4 shows treatment compliance of the trial patients. There were no significant interaction terms or between-group differences for compliances. When compliances were compared at each visit, there were no overall between-group differences among the two treatment groups.
Table 3 Demographic and baseline characteristics of the trial subjects
[TABLE="class: ivytablelight"]
<tbody>[TR]
[TH="colspan: 2"][/TH]
[TH]UC-II (N=26)[/TH]
[TH]G + C (N=26)[/TH]
[/TR]
[TR]
[TD="colspan: 2"]Age (years)[/TD]
[TD]58.9 ± 9.79[/TD]
[TD]58.7 ± 10.3[/TD]
[/TR]
[TR]
[TD="colspan: 2"]Sex: male/female (%)[/TD]
[TD]13/26 (50%)[/TD]
[TD]17/26 (65%)[/TD]
[/TR]
[TR]
[TD="colspan: 2"]Height (cm)[/TD]
[TD]167.7 ± 9.90[/TD]
[TD]167.0 ± 8.73[/TD]
[/TR]
[TR]
[TD="colspan: 2"]Weight (kg)[/TD]
[TD]84.3 ± 17.4[/TD]
[TD]86.6 ± 21.0[/TD]
[/TR]
[TR]
[TD="colspan: 2"]Systolic Blood Pressure (mm)[/TD]
[TD]128.2 ± 9.36[/TD]
[TD]126.3 ± 12.5[/TD]
[/TR]
[TR]
[TD="colspan: 2"]Diastolic Blood Pressure (mm)[/TD]
[TD]81.9 ± 7.43[/TD]
[TD]79.7 ± 8.60[/TD]
[/TR]
[TR]
[TD="colspan: 2"]Heart Rate (bpm)[/TD]
[TD]68.2 ± 7.72[/TD]
[TD]67.4 ± 8.47[/TD]
[/TR]
[TR]
[TD="colspan: 2"]Target knee[/TD]
[TD][/TD]
[TD][/TD]
[/TR]
[TR]
[TD][/TD]
[TD]Left; n (%)[/TD]
[TD]16 (61.5%)[/TD]
[TD]13 (50%)[/TD]
[/TR]
[TR]
[TD]Right; n (%)[/TD]
[TD]10 (38.5%)[/TD]
[TD]13 (50%)[/TD]
[/TR]
[TR]
[TD="colspan: 4"]Where applicable, values are expressed as mean ± SD[/TD]
[/TR]
</tbody>[/TABLE]
Table 4 Treatment compliance as assessed during specified visits
[TABLE="class: ivytablelight"]
<tbody>[TR]
[TH]Visit[/TH]
[TH="colspan: 2"]Treatment Group[/TH]
[/TR]
[TR]
[TH]UC-II[/TH]
[TH]G + C[/TH]
[/TR]
[TR]
[TD="colspan: 3"]AM Capsule Compliance[/TD]
[/TR]
[TR]
[TD]Visit 3[/TD]
[TD][25] 90.5 ± 19.2[/TD]
[TD][25] 93.6 ± 11.5[/TD]
[/TR]
[TR]
[TD]Visit 4[/TD]
[TD][24] 93.2 ± 9.66[/TD]
[TD][26] 94.5 ± 11.8[/TD]
[/TR]
[TR]
[TD]Visit 5[/TD]
[TD][23] 98.5 ± 5.15[/TD]
[TD][26] 93.3 ± 11.0[/TD]
[/TR]
[TR]
[TD="colspan: 3"]PM Capsule Compliance[/TD]
[/TR]
[TR]
[TD]Visit 3[/TD]
[TD][25] 88.1 ± 18.7[/TD]
[TD][25] 92.5 ± 12.5[/TD]
[/TR]
[TR]
[TD]Visit 4[/TD]
[TD][24] 92.8 ± 8.97[/TD]
[TD][26] 91.6 ± 12.3[/TD]
[/TR]
[TR]
[TD]Visit 5[/TD]
[TD][22] 95.3 ± 9.92[/TD]
[TD][26] 89.7 ± 12.6[/TD]
[/TR]
</tbody>[/TABLE]
There were no significant interaction terms and between-group differences for compliances. When compliances were compared at each visit, there were no overall between-group differences among the five treatment groups. Values are expressed as [n] mean ± SD.
WOMAC Score
The interaction between visit and treatment was significant in UC-II treated group for "pain walking on flat surface" (p=0.034), "difficulty walking on flat surface" (p=0.038) and "performing heavy domestic duties" (p=0.031) as compared to G+C treated group. There was evidence that UC-II treatment has a significant effect for “ascending stairs” (p=0.013) as compared to G+C treatment. Additionally, when groups were compared at each visit, UC-II was significantly better than G+C for “ascending stairs at 30 days and 60 days” (p=0.019 & 0.040 respectively), “at night while in bed” (p=0.015) at 60 days and difficulty walking on flat surface at 90 days (p=0.035). There were no further statistically significant differences for any other individual WOMAC components or summary scores. Treatment with UC-II was most effective and reduced the WOMAC scores by 33% compared to 14% in (G+C)-treated groups after 90 days. Within-group analysis indicated that treatment with UC-II for 90 days significantly (p<0.05) improved WOMAC scores at all treatment time points measured. In contrast, subjects received G+C did not show any statistical significant change in WOMAC scores at Day 90 of treatment (Fig. 2).
VAS Score
The interaction between visit and treatment was non-significant for all VAS components and summary scores. However there was evidence that UC-II treatment had a significant effect for “pain during climbing up and down stairs”, “night pain” and “resting pain” (p=0.035, 0.030 and 0.024 respectively). When groups were compared at each visit, UC-II was significantly better than G+C for “night pain” (p=0.040) and “resting pain” (p=0.020) at 60 days and “pain during climbing up and down stairs” (p=0.014) and “resting pain” at 90 days (p=0.034). There were no between-group differences for any of the VAS components or summary scores. Although both the treatments reduced the VAS score, UC-II was found to be more effective with a 40% decrease after 90 days of treatment compared to a 15% decrease in G+C treated groups.
Within-group analysis indicated that subjects on UC-II showed a significant reduction in total VAS scores at Day 60 and Day 90 as compared to baseline. However, subjects on G+C showed a significant reduction in total VAS scores at Day 30 and no significant difference was observed at either Day 60 or Day 90 as compared to baseline (Fig. 3).
Figure 2 Changes in WOMAC scores at Day 90 from baseline. WOMAC scores from each treatment group were compared to baseline value at specified time points. Each bar presents mean ± SEM. *p<0.05, **p<0.005 indicate significantly different from baseline.
 (Click on the image to enlarge.)
(Click on the image to enlarge.)Figure 3 Changes in VAS score at Day 90 from baseline. VAS scores from each treatment group were compared to baseline value at specified time points. Each bar presents mean ± SEM. **p<0.05 indicates significantly different from baseline.
 (Click on the image to enlarge.)
(Click on the image to enlarge.)Lequesne Score
The Lequesne's functional index was used to determine the effect of different treatments on pain during daily activities. The interaction between visit and treatment was non-significant for all Lequesne's components and summary scores. Furthermore, there were no between-group differences for any of the Lequesne's components or summary scores. However there was evidence that visit has a significant effect in UC-II treated group for “pain while up from sitting” and “maximum distance walked” (p=0.036 and 0.002 respectively) as compared to G+C treated group. There was as a strong trend toward UC-II efficacy. UC-II treatment effectively reduced Lequesne's functional index score by 20.1% as compared to 5.9 % by G+C treatment.
Within-group analysis suggested that subjects on UC-II demonstrated a significant reduction in total Lequesne's index of severity score from baseline to Day 90, whereas no significant difference from baseline was observed for subjects on G+C at any treatment time points evaluated (Fig. 4).
Figure 4 Changes in Lequesne's functional index at Day 90 from baseline. Lequesne's functional index from each treatment group was compared to baseline value at specified time points. Each bar presents mean ± SEM. *p<0.05 indicates significantly different from baseline.
 (Click on the image to enlarge.)
(Click on the image to enlarge.)Adverse Events
Adverse effects that occurred during the 90-day trial period are summarized in Table 5. Overall, there were 58 adverse events noted in the subjects receiving G+C treatment, whereas, only 35 adverse events were observed in UC-II group. In terms of severity, 60% of mild and 38% of moderate adverse events were experienced by subjects on G+C in comparison to 43% and 54% by subjects on UC-II. In relationship to test product a higher number of subjects (23%) on G+C demonstrated adverse events possibly related to product as compared to 11.4% of subjects on UC-II. For UC-II the possible adverse events related to products were constipation and headaches (intermittently). For G+C the possible adverse events related to products were bloating, stomach pain, rash, water retention (edema around eyes and scars), hives on face and chest, and headache. However, there was no significant difference in the occurrence of adverse effects between the two treatment groups.
Rescue Medication
A greater percentage of subjects used rescue medication while on G+C as compared to UC-II at every time point assessed. From baseline to Day 30 a total of 8 subjects (33.3%) on UC-II used rescue medication as compared to 23 subjects (88.5%) on G+C. From Day 30 to Day 60, 13 subjects (54.2%) on UC-II used rescue medication as compared to 21 subjects (80.8%) on G+C. Fourteen subjects (63.6%) on UC-II used rescue medication as compared to 19 subjects (79.2%) on G+C from Day 60 to Day 90.
Table 5 Summary of analysis of adverse events in all subjects
[TABLE="class: ivytablelight"]
<tbody>[TR]
[TH][/TH]
[TH="colspan: 2"]Treatment Group[/TH]
[/TR]
[TR]
[TH]UC-II (n=26)[/TH]
[TH]G + C (n=26)[/TH]
[/TR]
[TR]
[TD="colspan: 3"]Severity
[/TD][/TR]
[TR]
[TD]Mild[/TD]
[TD]15[/TD]
[TD]35[/TD]
[/TR]
[TR]
[TD]Moderate[/TD]
[TD]19[/TD]
[TD]22[/TD]
[/TR]
[TR]
[TD]Severe[/TD]
[TD]1[/TD]
[TD]1[/TD]
[/TR]
[TR]
[TD="colspan: 3"]Relationship to Test Article
[/TD][/TR]
[TR]
[TD]Not related[/TD]
[TD]17[/TD]
[TD]20[/TD]
[/TR]
[TR]
[TD]Unlikely[/TD]
[TD]14[/TD]
[TD]30[/TD]
[/TR]
[TR]
[TD]Possible[/TD]
[TD]4[/TD]
[TD]8[/TD]
[/TR]
[TR]
[TD]Probable[/TD]
[TD]0[/TD]
[TD]0[/TD]
[/TR]
[TR]
[TD]Most Probable[/TD]
[TD]0[/TD]
[TD]0[/TD]
[/TR]
[TR]
[TD="colspan: 3"]Body System
[/TD][/TR]
[TR]
[TD]Pain[/TD]
[TD]10[/TD]
[TD]17[/TD]
[/TR]
[TR]
[TD]Gastrointestinal[/TD]
[TD]5[/TD]
[TD]15[/TD]
[/TR]
[TR]
[TD]Musculoskeletal/Soft Tissue[/TD]
[TD]7[/TD]
[TD]5[/TD]
[/TR]
[TR]
[TD]Neurology[/TD]
[TD]0[/TD]
[TD]2[/TD]
[/TR]
[TR]
[TD]Pulmonary / Upper Respiratory[/TD]
[TD]2[/TD]
[TD]1[/TD]
[/TR]
[TR]
[TD]Hemorrhage/Bleeding[/TD]
[TD]2[/TD]
[TD]1[/TD]
[/TR]
[TR]
[TD]Blood/Bone Marrow[/TD]
[TD]2[/TD]
[TD]1[/TD]
[/TR]
[TR]
[TD]Dermatology/Skin[/TD]
[TD]2[/TD]
[TD]3[/TD]
[/TR]
[TR]
[TD]Allergy / Immunology[/TD]
[TD]0[/TD]
[TD]1[/TD]
[/TR]
[TR]
[TD]Infection[/TD]
[TD]1[/TD]
[TD]3[/TD]
[/TR]
[TR]
[TD]Lymphatics[/TD]
[TD]0[/TD]
[TD]1[/TD]
[/TR]
[TR]
[TD]Hepatobilary / Pancreatic[/TD]
[TD]0[/TD]
[TD]0[/TD]
[/TR]
[TR]
[TD]Renal / Genitoruinary[/TD]
[TD]0[/TD]
[TD]0[/TD]
[/TR]
[TR]
[TD]Constitutional Symptoms[/TD]
[TD]2[/TD]
[TD]3[/TD]
[/TR]
[TR]
[TD]Syndromes[/TD]
[TD]1[/TD]
[TD]1[/TD]
[/TR]
[TR]
[TD]Auditory/Ear[/TD]
[TD]0[/TD]
[TD]1[/TD]
[/TR]
[TR]
[TD]Ocular / Visual[/TD]
[TD]0[/TD]
[TD]1[/TD]
[/TR]
[TR]
[TD]Metabolic / Laboratory[/TD]
[TD]1[/TD]
[TD]2[/TD]
[/TR]
[TR]
[TD]Total Number of Adverse Events Experienced During Study
[/TD][TD]35[/TD]
[TD]58[/TD]
[/TR]
[TR]
[TD]Total Number of Subjects Experiencing Adverse Events: n (%)[/TD]
[TD]16/26 (61.5%)[/TD]
[TD]20/26 (76.9%)[/TD]
[/TR]
</tbody>[/TABLE]
see part 2
